We are unbelievably excited this week to be reviewing the hot-off-the-presses 2026 Multi-Society (AHA/ACC/ACCP/ACEP/CHEST/SCAI/SHM/SIR/SVM/SVN) Pulmonary Embolism Guidelines with lead author Dr. Mark A. Creager. We will talk about key updates in these guidelines compared to prior practice, including the new risk classification model, and provide an overview from diagnosis to follow-up. Given the clinical importance and prevalence of pulmonary embolism, these guidelines are certainly going to shape practice going forward, so this episode is a can’t miss!
Watch the full video of this episode with graphics and helpful teaching visuals on our YouTube channel: https://www.youtube.com/@pulmpeeps
Meet Our Guest
Dr. Mark Creager is a Professor of Medicine at Dartmouth Hitchcock Medical Center where he specializes in Cardiovascular Medicine with an emphasis on venous thromboembolic disease. He served as the lead author of the 2026 Pulmonary Embolism Guidelines.
This is the first joint AHA/ACC clinical practice guideline specifically on acute PE, bringing together a truly multidisciplinary writing committee (cardiology, pulmonology, hematology, emergency medicine, interventional radiology, surgery, and others). Prior guidelines existed from individual societies, but nothing this comprehensive had been updated in roughly five to six years.
New PE clinical categories (A through E):
One of the most impactful changes is replacing the old “massive/submassive” and “low/intermediate/high risk” labels with five categories that form a severity continuum. Category A is subclinical (incidental PE found on imaging in asymptomatic patients). Category B covers symptomatic but low-severity patients. Category C is where much of the clinical complexity lives — symptomatic, hemodynamically stable patients subdivided into C1, C2, and C3 based on RV function and biomarkers. Category D represents incipient cardiopulmonary failure (transient hypotension, normotensive shock with end-organ dysfunction). Category E is frank cardiopulmonary failure, with E2 being the sickest — refractory or recurrent cardiac arrest. Respiratory modifiers (hypoxia requiring supplemental oxygen) layer onto C, D, and E.
Diagnostic approach:
Clinical evaluation comes first — history, exam, and validated decision tools (Wells score, revised Geneva, PERC). If clinical probability is low and D-dimer is normal, imaging can be safely avoided. If either is concerning, imaging is warranted. CTPA remains the preferred imaging modality due to superior sensitivity, specificity, wide availability, and ability to assess clot burden and alternative diagnoses. VQ scanning is still appropriate when CTPA is contraindicated, and VQ SPECT offers better reproducibility and specificity than traditional planar VQ if available. Echocardiography is not a diagnostic test for PE but is important for risk stratification — RV size, TAPSE, and tissue Doppler measures all contribute prognostic information.
Anticoagulation updates:
Anticoagulation remains the cornerstone of treatment. For patients potentially needing advanced therapies (C3, D, E), parenteral anticoagulation is started first. A notable recommendation: low molecular weight heparin is generally preferred over unfractionated heparin, based on evidence showing more effective VTE risk reduction, more predictable pharmacokinetics, no need for routine monitoring, lower rates of heparin-induced thrombocytopenia, and no increase in major bleeding. The committee acknowledged this may create discomfort for clinicians accustomed to unfractionated heparin’s easy reversibility, but the difficulty of achieving and maintaining therapeutic levels with UFH was a significant concern.
Advanced therapies:
Catheter-based thrombolysis, mechanical thrombectomy, systemic thrombolysis, and surgical embolectomy all received mostly class 2B recommendations (“can consider”) for C3 and D categories, reflecting that current evidence shows improvement in short-term surrogate measures (RV/LV ratio, hemodynamics) but lacks definitive hard outcome data on mortality. For category E1 patients, recommendations are stronger (class 2A). Multiple trials are expected soon — HI-PEITHO, PEERLESS-2, PE-TRACT, PERSEVERE, TORPEDO, and PROG — that should substantially inform future updates.
PERT teams:
Pulmonary embolism response teams are encouraged, particularly for C3, D, and E patients. They’ve been shown to reduce length of stay. For institutions without PERT capability, establishing consultation networks with larger centers is recommended.
Post-PE follow-up:
Patients shouldn’t be “left in the wilderness” after discharge. The guidelines recommend communication within the first week to ensure understanding of diagnosis and treatment, an in-person visit at or before three months to assess for persistent symptoms and discuss anticoagulation duration, ongoing surveillance for chronic thromboembolic pulmonary disease, and periodic reassessment for those on extended anticoagulation.
On this week’s episode, we’re continuing our Guidelines Series exploring the 2022 ESC/ERS Guidelines for the diagnosis and treatment of Pulmonary Hypertension. If you missed our first episode in the series, give it a listen to hear about the most recent recommendations regarding Pulmonary Hypertension definitions, screening, and diagnostics. Today, we’re talking about the next steps after diagnosis. Specifically, we’ll be discussing risk stratification, establishing treatment goals, and metrics for re-evaluation. We’ll additionally introduce the mainstays of pharmacologic therapy for Pulmonary Hypertension.
Meet Our Co-Hosts
Rupali Sood grew up in Las Vegas, Nevada and made her way over to Baltimore for medical school at Johns Hopkins. She then completed her internal medicine residency training at Massachusetts General Hospital before returning back to Johns Hopkins, where she is currently a pulmonary and critical care medicine fellow. Rupali’s interests include interstitial lung disease, particularly as related to oncologic drugs, and bedside medical education.
Tom Di Vitantonio is originally from New Jersey and attended medical school at Rutgers, New Jersey Medical School in Newark. He then completed his internal medicine residency at Weill Cornell, where he also served as a chief resident. He currently is a pulmonary and critical care medicine fellow at Johns Hopkins, and he’s passionate about caring for critically ill patients, how we approach the management of pulmonary embolism, and also about medical education of trainees to help them be more confident and patient centered.
Key Learning Points
1) Episode Roadmap
How to set treatment goals, assess symptom burden, and risk-stratify patients with suspected/confirmed pulmonary arterial hypertension (PAH).
What tools to use to re-evaluate patients on treatment
Intro to major PAH medication classes and how they map to pathways.
2) Case-based diagnostic reasoning
Patient: 37-year-old woman with exertional dyspnea, mild edema, abnormal echo, telangiectasias + epistaxis → raises suspicion for HHT (hereditary hemorrhagic telangiectasia) and/or early connective tissue disease.
Key reasoning move: start broad (Groups 2–5) and narrow using history/exam/testing.
In a young patient without obvious left heart or lung disease, think more about Group 1 PAH (idiopathic/heritable/associated).
HHT teaching point: HHT can cause PH in more than one way:
More common: high-output PH from AVMs (often hepatic/pulmonary)
Rare (1–2% mentioned): true PAH phenotype (vascular remodeling; associated with ALK1 in some patients), behaving like Group 1 PAH.
3) Functional class assessment
WHO Functional Class:
Class I: no symptoms with ordinary activity, only with exertion
Class II: symptoms with ordinary activity
Class III: symptoms with less-than-ordinary activity (can’t do usual chores/shopping without dyspnea)
Class IV: symptoms at rest
Practical bedside tip they give:
Ask if the patient can walk at their own pace or keep up with a similar-age peer/partner. If not, think Class II (or worse).
4) Risk stratification at diagnosis: why, how, and which tools
Big principle: treatment choices are driven by risk, and the goal is to move patients to low-risk quickly.
Clinical progression, signs of right heart failure, syncope
WHO FC
Biomarkers (NT-proBNP)
Exercise capacity (6MWD)
Hemodynamics
Imaging (echo; sometimes cardiac MRI)
CPET (peak VO₂; VE/VCO₂ slope)
They note: even if you don’t have everything, the calculator can still be useful with ≥3 variables.
REVEAL 2.0:
Builds on similar core variables but adds further patient context (demographics, renal function, BP, DLCO, etc.)
Case result: both tools put her in intermediate risk (ESC/ERS ~1.6; REVEAL 2.0 score 8), underscoring that mild symptoms can still equal meaningful mortality risk.
5) Treatment goals and follow-up philosophy
What they explicitly prioritize:
Help patients feel better, live longer, and stay out of the hospital
Use risk tools to communicate prognosis and to track improvement
Reassess frequently (they mention ~every 3 months early on) until low risk is achieved
“Time-to-low-risk” is an important treatment goal
Also emphasized:
The diagnosis is psychologically heavy; patients need clear counseling, reassurance about the plan, and connection to support groups.
6) Medication classes for the treatment of PAH
Nitric oxide–cGMP pathway
PDE5 inhibitors: sildenafil, tadalafil
Soluble guanylate cyclase stimulator: riociguat
Important safety point: don’t combine PDE5 inhibitors with riociguat (risk of significant hypotension/hemodynamic effects)
Endothelin receptor antagonists (ERAs)
“-sentan” drugs: bosentan (less used due to side effects/interactions), ambrisentan, macitentan
Teratogenicity emphasized
Hepatotoxicity that requires LFT monitoring
Can cause fluid retention and peripheral edema
Prostacyclin pathway
Prostacyclin analogs/agonists:
Epoprostenol (potent; short half-life; IV administration)
Treprostinil (IV/SubQ/oral/inhaled options)
Selexipag (oral prostacyclin receptor agonist)
7) Sotatercept (post-guidelines)
They note sotatercept wasn’t in 2022 ESC/ERS but is now “a game changer” in practice:
Positioned as potentially more disease-modifying than pure vasodilators
Still evolving: where to place it earlier vs later in regimens is an active question in the field
8) How risk category maps to initial treatment intensity
General approach they outline:
High risk at diagnosis: parenteral prostacyclin (IV/SubQ) strongly favored, often aggressive early
Intermediate risk: at least dual oral therapy (typically PDE5i + ERA); escalate if not achieving low risk
Low risk: at least one oral agent; many still use dual oral depending on etiology/trajectory
For the case: intermediate-risk → start dual oral therapy (they mention tadalafil + ambrisentan as a typical choice), reassess in ~3 months; add a third agent (e.g., selexipag/prostacyclin pathway) if not low risk.
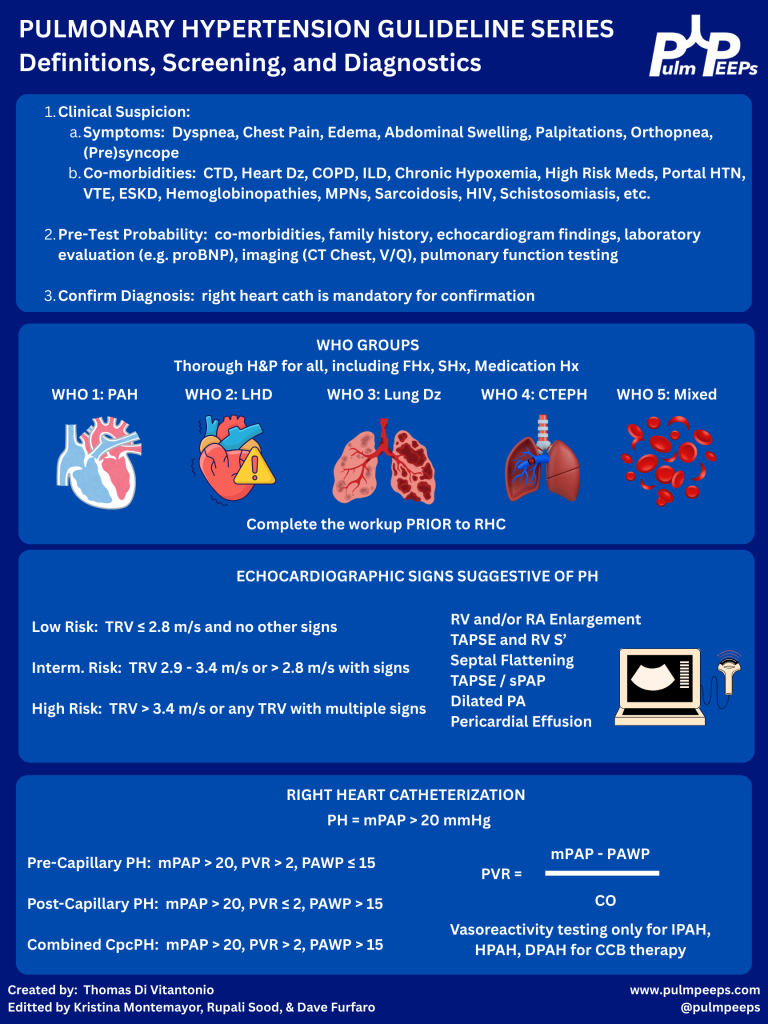
Today we’re kicking off another segment in our Guidelines Series, and doing a deep dive into the 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Over a series of episodes we’ll talk about the most recent updates to definitions around pulmonary hypertension, recognizing and diagnosing Group 1 – 5 pulmonary hypertension, risk stratification, and treatments. In this first episode, we will review the most recent definitions, including changes to the definitions that were new in 2022. We’ll then talk about recognizing and diagnosing pulmonary hypertension with tips and insights along the way.
Meet Our Co-Hosts
Rupali Sood grew up in Las Vegas, Nevada and made her way over to Baltimore for medical school at Johns Hopkins. She then completed her internal medicine residency training at Massachusetts General Hospital before returning back to Johns Hopkins, where she is currently a pulmonary and critical care medicine fellow alongside Tom. Rupali’s interests include interstitial lung disease, particularly as related to oncologic drugs. And she also loves bedside medical education.
Tom Di Vitantonio is originally from New Jersey and attended medical school at Rutgers, New Jersey Medical School in Newark. He then completed his internal medicine residency at Weill Cornell, where he also served as a chief resident. He currently is a pulmonary and critical care medicine fellow at Johns Hopkins, and he’s passionate about caring for critically ill patients, how we approach the management of pulmonary embolism, and also about medical education of trainees to help them be more confident and patient centered in the care they have going forward.
Infographic
Key Learning Points
Why to have a high index of suspicion for pulmonary hypertension (PH)
PH often presents subtly with slowly progressive dyspnea on exertion, fatigue, lightheadedness, exertional chest pain, or syncope.
There’s often a delay of 1–2+ years from symptom onset to diagnosis, which is associated with worse mortality.
Early recognition and treatment, especially for pulmonary arterial hypertension (PAH, WHO group 1), can significantly change outcomes.
When to suspect PH
Think PH when:
Dyspnea is out of proportion to:
CT parenchymal findings (relatively normal lungs)
Spirometry (normal FEV₁/FVC, volumes)
There are subtle but progressive symptoms over months:
Reduced exercise tolerance
No obvious alternative explanation (e.g., no overt HF, CAD, big ILD, etc.)
Physical exam may show (often late):
Elevated JVP, V waves (TR)
Peripheral edema, hepatomegaly, ascites
Loud P2, RV heave
In the case: a woman with systemic sclerosis + slowly progressive exertional dyspnea + relatively normal CT parenchyma and spirometry → high suspicion.
WHO classification: 5 PH groups (big picture + why it matters)
Used for pathophysiology, prognosis, and treatment choices:
We’re back with our Rapid Fire Journal Club, and talking about the NEJM 2023 STELLAR Trial of Sotatercept in Pulmonary Arterial Hypertension. This is a landmark trial that is actively changing the face of PAH treatment today. Listen to hear the details of the trial and how its findings can be utilized to help patients.
Article and Reference
We’re looking at the STELLAR Trial today which is a Phase 3 trial of Sotatercept in Pulmonary Arterial Hypertension.
Listen in today to another stop on our Fellows’ Case Files journey. We’re at Northwestern University for another great case presentation. Tune in, check out our associated infographic, and let us know what you think!
Meet Our Guests
Jamie Rowell is a first-year clinical fellow in the Northwestern PCCM program. She completed medical school at the Medical University of South Carolina and her internal medicine residency and Chief Residency at the University of Vermont Medical Center.
Cathy Gao is an Instructor of Medicine at Northwestern and completed her PCCM fellowship there last year. Her research focuses on using machine learning applied to ICU EHR data to characterize patient trajectories and identify potential interventions to improve outcomes.
Clara Schroedl is an Associate Professor of Medicine in Pulmonary and Critical Care and Medical Education. She is the program director of the Northwestern PCCM fellowship program, with an interest in medical education and simulation.
Case Presentation
A 25-year-old previously healthy woman presents with recurrent episodes of right chest pain and cough. In October she was treated with antibiotics and felt somewhat better but in December, she presented again with chest pain, and again was treated with antibiotics. The pain improved but she still felt breathless. In February, again she had intense chest pain interfering with life, and was given NSAIDs and took high dose TID without clear benefit.
One month later, she coughed up some bloody mucus, so now she is presenting for evaluation. The chest pain is worse with deep breaths and improves in between these episodes. She only notes it on her right side. At this point, she does sometimes feel short of breath; she used to run 5 miles but is now struggling to run two miles. She denies any unusual exposures. She went to school in central rural Ohio for a while. She has no history of pulmonary infections, no exposure to mold or animals, and no history of vaping.
Key Learning Points
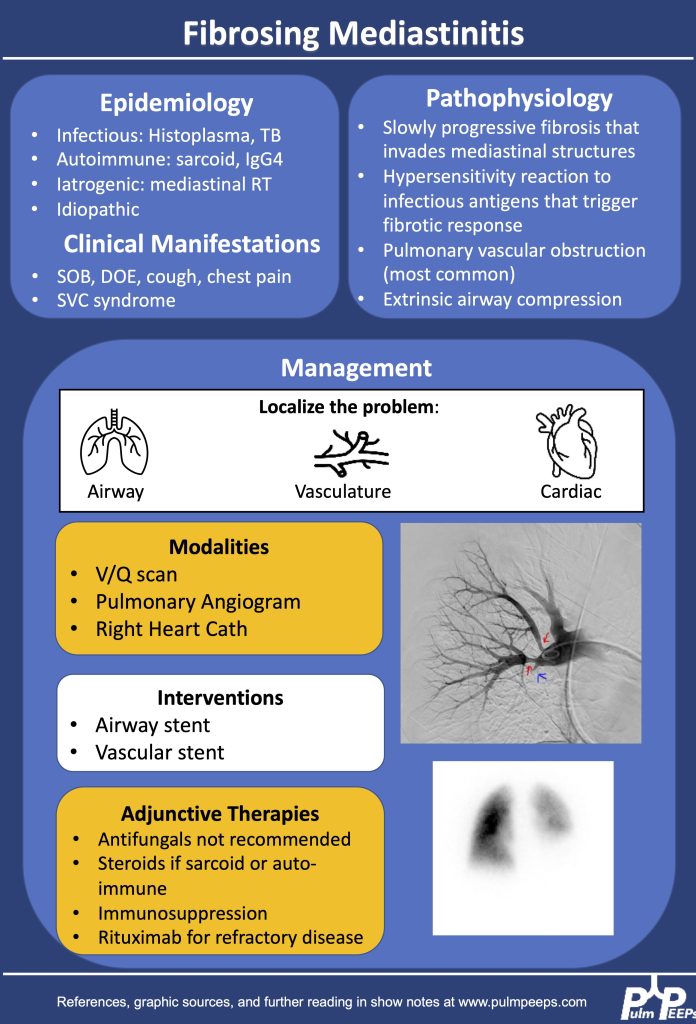
1.Making the diagnosis of Fibrosing Mediastinitis :
–Etiologies: histoplasmosis, sarcoidosis, tuberculosis, IgG4, Behcet, ANCA vasculitis
–Imaging characteristics: infiltrative, heterogeneous, fibrotic process that crosses fat planes and encroaches on nearby structures causing airway or vascular stenoses
2. Management strategies:
–No curative therapies. Goal to relieve symptom burden
–Airway stents
–Vascular stents
–Rituximab
–Antifungals, steroids generally not considered effective
References and Further Reading
Kern et al. Bronchoscopic Management of Airway Compression due to Fibrosing Mediastinitis. Annals of the American Thoracic Society 2017. 14: 1235-1359
Welby JP, Fender EA, Peikert T, Holmes DR Jr, Bjarnason H, Knavel-Koepsel EM. Evaluation of Outcomes Following Pulmonary Artery Stenting in Fibrosing Mediastinitis. Cardiovasc Intervent Radiol. 2021 Mar;44(3):384-391. doi: 10.1007/s00270-020-02714-z. Epub 2020 Nov 17. PMID: 33205295.
Westerly, BD Targeting B Lymphocytes in Progressive Fibrosing Mediastinitis.Am J Respir Crit Care Med. 2014 Nov 1; 190(9): 1069–1071.
We’re back with our Top Consults series to talk about Lung Transplant! This is a topic that every pulmonologist should have background knowledge about since it impacts the care of patients with end-stage lung disease of any cause. We will talk about the indications for referral and transplant, how to advise patients and some unique considerations for evaluation. Enjoy, rate and review us, and share your thoughts about the episode!
Meet Our Guests
Dr. Meghan Aversa is an Assistant Professor of Medicine at the University of Toronto and her expertise involves patients with end stage lung disease and lung transplant.
Dr. Hannah Mannemis an Associate Professor of Medicine at the University of Virginia Health. Hannah joined faculty at UVA in 2016 and she has expertise in ILD and Lung Transplant.
Learning Points
Trends in lung transplant:
Global Increase in Lung Transplants: Over the past three decades, there has been a gradual worldwide increase in lung transplants, with approximately 4,500 performed annually. North America conducts over half of these transplants, and the growth is particularly notable in double lung transplants.
Indications and Disease Trends: Interstitial lung disease (ILD) has seen a significant rise in lung transplant indications, surpassing COPD as the leading cause. ILD, especially idiopathic pulmonary fibrosis (IPF), constitutes a substantial portion (40%) of all transplants. However, the trend is primarily observed in North America.
Decline in Cystic Fibrosis Cases: While Cystic Fibrosis is still a significant indication for lung transplant, its percentage has been declining, likely due to improvements in drugs and CFTR modulators.
Evolution of Lung Transplant Candidates: Over the past five years, lung transplant candidates have become sicker, with higher listing scores and increased hospitalization rates at the time of transplant. More patients have antibodies affecting match difficulty. The average age of patients has increased, with 35% being over 65, a demographic that was previously considered contraindicated.
Impact of COVID-19: The COVID-19 pandemic has influenced lung transplant trends. In 2020, UNOS added COVID-19-related ARDS and pulmonary fibrosis as indications. In 2021, these indications constituted about 10% of lung transplants, making it the third most common indication. Two-thirds were due to COVID-19 ARDS, and one-third due to pulmonary fibrosis. The long-term impact, especially with evolving vaccine dynamics, is still uncertain.
Indications for transplant referral:
ISHLT Consensus Document Update (2021): The ISHLT consensus document for lung transplant candidate selection was updated in 2021. It is available on the ISHLT website and serves as a valuable guideline for pulmonologists considering referrals for lung transplant assessment.
General Rule of Thumb for Chronic Lung Diseases: According to the consensus document, a general rule of thumb for all patients with chronic and stage lung diseases is to consider lung transplant if there is a high (more than 50%) risk of death from the lung disease within the next two years. Prognostic markers vary based on the underlying lung disease.
Disease-Specific Recommendations: The consensus document provides disease-specific recommendations. The key diseases highlighted are COPD, ILD, CF, and PH.
COPD: Referral is recommended when the BODE index is in the range of 5 to 6, with additional factors that increase mortality, such as frequent exacerbations, low FEV1 (20-25%), or rapidly increasing BODE. Referral is also advised for clinically deteriorating patients or those with an unacceptably low quality of life despite maximal medical therapy.
ILD (Particularly IPF): Early referral is suggested, ideally at the time of diagnosis. For any pulmonary fibrosis, referral is recommended if FEC is less than 80% or declining by 10% in two years, or DLCO is less than 40% or declining by 15% in two years. Other factors for referral include radiographic progression or a need for supplemental oxygen.
Cystic Fibrosis (CF): Referral is encouraged for those with FEV1 less than 30%, and even 40% if there’s reduced walk distance, hypercapnia, PH, frequent exacerbations, or rapid decline.
Pulmonary Hypertension (PH): Referral criteria include a REVEAL score of eight, significant RV dysfunction, progressive disease on therapy, need for IV prostacyclin therapy, and specific conditions like PVOD, PCH, scleroderma pulmonary artery aneurysms, which should be referred early due to their rapid progression.
Transplant evaluation process
Phases of Lung Transplant Evaluation:
Referral and Initial Visit: The process begins with a referral, often from a primary pulmonologist. Patients can also self-refer. The initial phase involves insurance authorization and confirming the underlying diagnosis while ensuring all other treatment options are exhausted.
Assessment of Disease Severity: The severity of end-stage lung disease is assessed to determine the timing of the workup, which varies depending on the patient’s condition and the center’s protocols.
Diagnostic Steps: A thorough diagnostic workup follows the initial visit, including various tests, imaging, and meetings with multidisciplinary teams to assess medical and social factors influencing transplant success.
Follow-Up Appointments: Patients typically have multiple follow-up appointments to track the evolution of the disease and ensure health maintenance and vaccinations are up to date.
Selection Committee: The final phase involves a selection committee that determines if the patient is a candidate. If so, there may be conditional requirements before officially listing the patient.
Multidisciplinary Approach: Lung transplant evaluation involves collaboration with various specialists, including social work, finance, nutrition, pharmacy, physical therapy, and potentially other consult services. The efficiency of this process is optimized for both the patient and the medical team.
Diagnostic Workup:
Medical Testing: Involves blood work, cardiac testing (echo, left and right heart cath), and imaging, including abdominal imaging, VQ scans, DEXA scans, and 24-hour urine analysis.
Multidisciplinary Meetings: Patients meet with members of the multidisciplinary team, addressing medical comorbidities as well as social and psychological factors.
Follow-Up Appointments: Multiple appointments allow for tracking disease progression and ensuring overall health maintenance.
Selection Committee Decision: The patient receives a decision from the selection committee, determining candidacy. Sometimes, patients are considered candidates with conditions (e.g., completing vaccinations or losing weight). Timing of listing is also discussed to ensure optimal candidacy.
Patient Involvement: Patients play an active role, and the process may involve self-referral, understanding and completing requirements, and active participation in follow-up appointments.
Efficiency and Individualization: The evaluation process is tailored to the patient’s condition, and centers aim to efficiently organize diagnostic workup and multidisciplinary meetings to optimize patient care.
Timing of transplant listing for candidates
COPD Patients: For COPD patients, listing is likely when the Bode index is around 7, the FEV1 is under 20%, there is at least moderate pulmonary hypertension (PH), chronic hypercapnia, or severe exacerbations.
ILD Patients: Patients with interstitial lung disease (ILD) are likely to be listed when showing signs of progression or decline in forced expiratory capacity (FEC), diffusing capacity of the lungs for carbon monoxide (DLCO), or six-minute walk distance. Other indicators include hypoxemia, secondary pulmonary hypertension, or hospitalization for complications.
CF Patients: Cystic fibrosis (CF) patients are considered for listing when FEV1 is below 25% or is rapidly declining, and if they experience frequent hospitalizations. Listing criteria also include the presence of pulmonary hypertension, chronic hypoxemia, or hypercapnia.
Pulmonary Hypertension Patients: Those with primary pulmonary hypertension may be listed when the reveal score is above 10 on intravenous therapy, there is progressive hypoxemia, or if there are renal or liver dysfunctions associated with pulmonary hypertension (PH).
Changes from the LAS system to the CAS system
Transition to Composite Allocation Score (CAS):
Background and Timing: In March 2023, the lung allocation system (LAS) transitioned to the composite allocation score (CAS), a major change in the allocation of lung transplants.
Reasoning Behind the Change: The change aimed to improve organ matching, prioritize sick candidates, enhance long-term survival, promote equity, increase transplant opportunities for specific patient groups (especially pediatric patients), and manage geographical variation in organ placement.
Components of CAS:
Medical Urgency: Based on waitlist mortality at one year without a transplant and the likelihood of survival post-transplant, now assessed at greater than five years, with equal weighting.
Recipient Variables: Includes factors like height discrepancy, blood type matching, sensitization (immune system matching), and other recipient variations.
Candidate Biology: Focuses on pediatric patients (less than 18 years old) and individuals are a prior living donor.
Donor Variables: Addresses donor characteristics, emphasizing proximity and travel distance from the organ hospital.
Early Data and Observations: The initial three-month monitoring period has shown changes in O blood type scores, prompting adjustments. Notable outcomes include a 16% increase in the number of lung transplants, a decrease in waitlist deaths and removals, and changes in median distance between donor hospital and transplant center.
Exception Scores: The number of exception scores has increased, allowing for adjustments when the assigned score may not reflect the patient’s true medical urgency.
Caution and Early Analysis: Early data, while promising, is subject to caution as centers were aware of the upcoming change. The impact on different age groups and the reasons for exceptions are being closely monitored and may evolve as more data becomes available.
Ongoing Monitoring and Potential Evolution: The data is being closely tracked by medical directors, and further changes to the scoring system may occur based on ongoing analysis and experience with the CAS. The impact on patient outcomes and allocation efficiency will continue to be studied and refined.
Advising patients on what to expect in terms of prognosis and survival after lung transplant
Survival Statistics:
Overall three is approximately 50 percent survival at five years, and the median survival time is approximately six and a half years.
Significant variations based on factors such as diagnosis, age, and comorbidities.
Survival outcomes differ for specific groups, e.g., cystic fibrosis (CF) patients, those older than 65, and individuals with interstitial lung disease (ILD).
Quality of Life Emphasis:
Shift in focus from survival alone to the patient’s goals and quality of life.
Highlighting the importance of understanding and aligning with the patient’s individual quality of life expectations.
Investment in Healthcare Team and Lifestyle Change:
Emphasis on the long-term commitment and involvement with the healthcare team post-transplant.
A substantial investment in healthcare post-transplant, including regular visits, extensive blood work, and medication management.
Cultural shift for patients to adapt to a new routine of frequent medical visits even when otherwise healthy.
Complications and Side Effects:
Acknowledgment of potential complications within the first year, making the initial post-transplant period a full-time job.
Discussion of various complications and medication side effects, ensuring patients are informed.
Multidisciplinary approach involving nutritionists, physical therapists, and other specialists to address complications and enhance the patient’s quality of life.
Individualized Patient Approach:
Recognition of the patient’s fight, spirit, and motivation as crucial factors for successful transplantation.
Encouraging patients to set goals for their post-transplant life.
Ethical considerations regarding transplanting older patients, with the importance of assessing overall well-being, motivation, and mental health.
Acknowledgment of Averages and Unpredictability:
Communication of averages, but a reminder of the inherent unpredictability in the post-transplant course.
Preparing patients for potential complications and the need to adapt to unforeseen challenges.
Managing expectations by highlighting the unpredictability of individual transplant journeys.
Quality of Life Improvement:
Despite complications and side effects, lung transplant often results in a significant improvement in the patient’s quality of life.
Patients generally experience increased satisfaction and happiness post-transplant, outweighing the challenges associated with the procedure and subsequent care.
We are extremely excited to be hosting this episode in collaboration with CardioNerds! We have known Amit and Dan for many years, and they have been huge supporters of Pulm PEEPs, so it is an honor to address a topic we’re all interested in together.
We are joined by experts in the field today to discuss acute, decompensated right ventricle failure in patients with Pulmonary Arterial Hypertension (PAH). This topic can be quite intimidating, so we hope this will serve as a valuable guide for anyone who encounters a patient like this in the ICU.
Meet Our Guests
Leonid “Leon” Mirson is an internal medicine resident at the Johns Hopkins Hospital Osler Medical Residency and an Associate Editor here at Pulm PEEPs. He was born in Ukraine and moved to Philadelphia in early childhood with his family. He received his undergraduate degree from the University of Pittsburgh where he studied biomedical engineering and received his medical degree from the University of Pittsburgh School of Medicine. His current interests include pulmonary and critical care medicine with a focus on pulmonary hypertension as well as medical education. He is a rising PCCM fellow at the University of Pennsylvania.
Bhavya Varma completed her medical school at the University of Pittsburgh, her internal medicine residency at Johns Hopkins, and is a rising Cardiology fellow at NYU. She is interested in medical education and has done work with CardioNerds during her residency.
Mardi Gomberg-Maitland is a Professor of Medicine at George Washington University. She serves as the Medical Director of the Pulmonary Hypertension Program at George Washington Hospital. She completed her medical degree at Albert Einstein College of Medicine, completed her residency at the Weill-Cornell Medical Center, and completed her fellowship in cardiovascular diseases at Mount Sinai Medical Center. Her research focus is on understanding the epidemiology of pulmonary hypertension and the development of novel therapeutics and biomarkers. Dr. Gomberg-Maitland is internationally known for her work, she has had extensive grant funding and has published over 150 articles, abstracts, reviews, and chapters.
Rachel Damico is a pulmonologist and an Associate Professor of Medicine at Johns Hopkins Hospital, where she is also the Associate Director of the physician-scientist training program. Dr. Damico received her medical degree and doctoral degree in Molecular and Cellular biology from the University of Pennsylvania. She completed her residency in the Osler Internal Medicine training program and continued on as a PCCM fellow at Johns Hopkins. She has quickly achieved an international reputation in the field of pulmonary vascular biology and both basic and translational research, as well as clinical excellence, in Pulmonary Arterial Hypertension.
Patient Presentation
A 21-year-old woman with a past medical history notable for congenital heart disease (primum ASD and sinus venosus with multiple surgeries) complicated by severe PAH on home oxygen, sildenafil, ambrisentan, and subcutaneous treprostinil is presenting with palpitations, chest pain, and syncope. She presented as a transfer from an outside ED where she arrived in an unknown tachyarrhythmia and had undergone DCCV due to tachycardia into the 200s and hypotension. On arrival at our hospital, she denied SOB but did endorse nausea, leg swelling, and poor medication adherence. Her initial vitals were notable for a BP of 80/50, HR 110, RR 25, and saturating 91% on 5L O2. On exam, she was uncomfortable appearing but mentating well. She had cool extremities with 1-2+ LE edema. Her JVP was 15cm H2O. She has an RV Heave and 2/6 systolic murmur. Her lungs were clear bilaterally. Her labs were notable for Cr 2.0, an anion gap metabolic acidosis (HCO3 = 11), elevated lactate (4.1), elevated troponin to 14, and a pro-BNP of ~5000. Her CBC was unremarkable. Her EKG demonstrated 2:1 atrial flutter at a rate of 130.
Key Learning Points
Diagnosing RV failure in patients with PH:
RV dysfunction and RV failure are two separate entities. RV dysfunction can be measured on echocardiography, but RV failure can be thought of as a clinical syndrome where there is evidence of RV dysfunction and elevated right sided filling pressures.
RV failure is a spectrum and can present with a range of manifestations from evidence of R sided volume overload and markers of organ dysfunction, all the way to frank cardiogenic shock. Most patients with RV failure are not in overt shock.
One of the first signs of impending shock in patients with RV failure is the development of new or worsening hypoxemia. Patients with decompensated RV failure approaching shock often do not present with symptoms classic for LV low flow state. Instead, hypoxia 2/2 VQ mismatching may be the first sign and they can be otherwise well appearing. Particularly because patients with PH tend to be younger, they can often appear compensated until they rapidly decompensate.
Causes of decompensation for patients with RV dysfunction and PH:
Iatrogenesis (inadvertent cessation of pulmonary vasodilators by providers, surgery if providers are not familiar with risks of anesthesia), non-adherence to pulmonary vasodilators (either due to affordability issues or other reasons), infections, arrhythmias (particularly atrial arrhythmias), and progression of underlying disease.
Patients with atrial arrhythmias (atrial flutter or atrial fibrillation) and pulmonary hypertension do not tolerate the loss of the atrial kick well as it contributes a significant amount to their RV filling and impacts their cardiac output. It is often difficult to determine if the atrial arrhythmia is a cause or effect of decompensated RV failure, but its presence is associated with a worse prognosis. Efforts should be made to re-establish normal sinus rhythm in patients with decompensated RV failure and atrial arrhythmias.
A patient’s home PH medications should never be stopped for any reason upon admission unless on the basis of recommendations by a pulmonary hypertension provider as this is often a cause of decompensation inpatient
Interpreting findings on echocardiogram:
Echo is a useful screening tool. When interpreting evidence of RV dysfunction, it is important to look at the global picture and not just one measurement.
RVSP, though commonly reported, may be of limited value when evaluating for decompensation. It’s a function of blood pressure, heart rate, and cardiac output. RVSP may even decline as shock worsens.
TAPSE is useful as a marker of RV dysfunction if it is reduced, but it is difficult to follow over time and only gives information about cardiac function around the annulus; it may be normal even when apical RV function is depressed. RV fractional area of change may be more useful for global RV function. It is important to pay attention to the RV size overall, the degree of TR, and the presence of effusion all of which are associated with RV dysfunction.
Tips regarding the interpretation of invasive hemodynamics:
Cardiac output by thermodilution is the standard way to calculate PVR. Despite the degree of TR that is typically present, it is thought to be a better representation of cardiac output than the estimated Fick calculation.
Our experts agree that routine monitoring of invasive hemodynamics for acute decompensated RV failure is likely not helpful and has significant risks. A good external volume exam or CVP off a central venous catheter + central venous saturation will likely be all you need to navigate a patient with shock secondary to RV failure. A right heart catheterization (should be only done under fluoroscopy for patients with large RVs) may be helpful if the etiology of shock is unclear.
Management of decompensated RV failure in patients with pulmonary hypertension
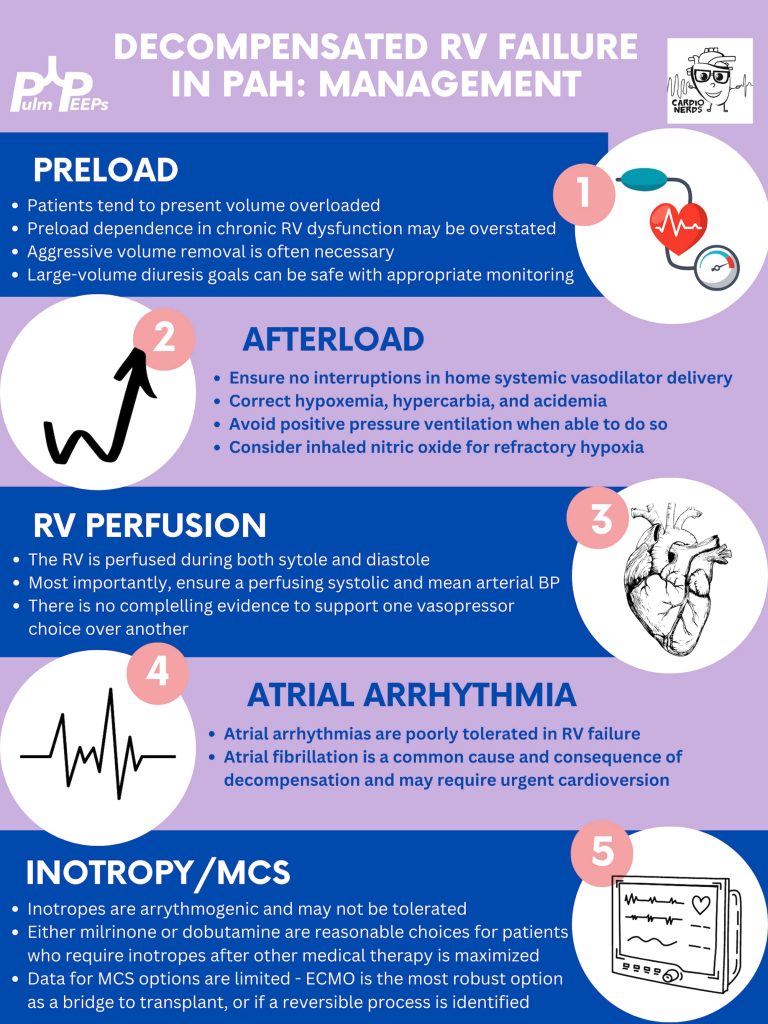
Managing preload is of utmost importance, perhaps the most important tenant of management of decompensated RV failure. The overwhelming majority of patients with PH and decompensation are volume overloaded, it is exceptionally rare that someone would be dry. Furthermore, the myth that the RV is “preload responsive” is only true in the setting of acute RV injury (eg. RV infarction) and not so in patients with acute on chronic RV dysfunction. It is important to optimize preload in someone in decompensated RV failure and it is safe to do this more rapidly than traditionally taught. Exact goals varied between our experts, but anywhere from 2-4L net negative per day is reasonable especially if the patient is hemodynamically tolerating the fluid removal. If the patient is not responding to diuretics, hemodialysis with ultrafiltration may be necessary to optimize the patient.
Afterload is the next tenant of management. Optimizing the following parameters will reduce the patient’s pulmonary vascular resistance and reduce afterload to the right ventricle.
— Avoiding hypoxic pulmonary vasoconstriction, liberalize the patient’s O2 goal
— Avoid permissive hypercapnia and academia in this patient population
— Do not withhold a patient’s pulmonary vasodilator until discussion with the PH team. If stopped inadvertently, restart this medication immediately. For patients with malfunctioning pumps, there is a phone number on the back that you can call for rapid troubleshooting. Sildanefil can be given IV if a patient is NPO.
— Inhaled nitric oxide can improve oxygenation and reduce afterload
— Intubation and mechanical ventilation greatly increase PVR and are poorly tolerated. Exacting care must be taken to titrate PEEP and tidal volume, and avoid intubation when possible.
— Starting a new systemic pulmonary vasodilator in decompensated RV failure may be considered under close guidance from the pulmonary hypertension team
Management of atrial arrhythmias:
As above, patients with severe pulmonary hypertension do not tolerate loss of sinus rhythm well. If they are decompensated, every effort should be made to re-establish normal sinus rhythm.
Management of RV perfusion:
Unlike the LV, the RV is perfused during BOTH systole and diastole. Maintaining effective coronary perfusion to the RV is essential in RV failure. For this reason, the systemic systolic pressure (as well as the mean arterial pressure) should be kept high enough to ensure that the RV is able to perfuse. There is no great body of evidence as to which pressor works best. Norepinephrine, vasopressin, and even phenylephrine are all reasonable choices to maintain appropriate perfusing blood pressure.
Inotropy:
Patients in shock and RV failure do not always require inotropes, but if they do it’s often a sign of a grim prognosis. Either dobutamine or milrinone is reasonable, but the negative effects of these drugs (arrhythmias, tachycardia, and systemic hypotension) may limit their uses.
Mechanical circulatory support:
Limited options are available. Balloon pumps and Impella devices have limited roles except in expert centers, and ECMO remains the standard of care. ECMO (either V-V or V-A) may have utility as a bridge to recovery if a reversible cause is identified, or a bridge to transplant if the patient is on the transplant list.
Goals of care:
The prognosis of a patient admitted to the ICU with acute on chronic decompensated RV failure is guarded, with very high mortality rates even if not in shock
It is important for the patient’s longitudinal pulmonary hypertension provider to discuss the prognosis and goals of care ahead of time but this is not always possible. If they are admitted, early discussions regarding code status and prognosis are essential. It may be helpful to bring in the patient’s longitudinal pulmonary hypertension doctor into these discussions if possible.
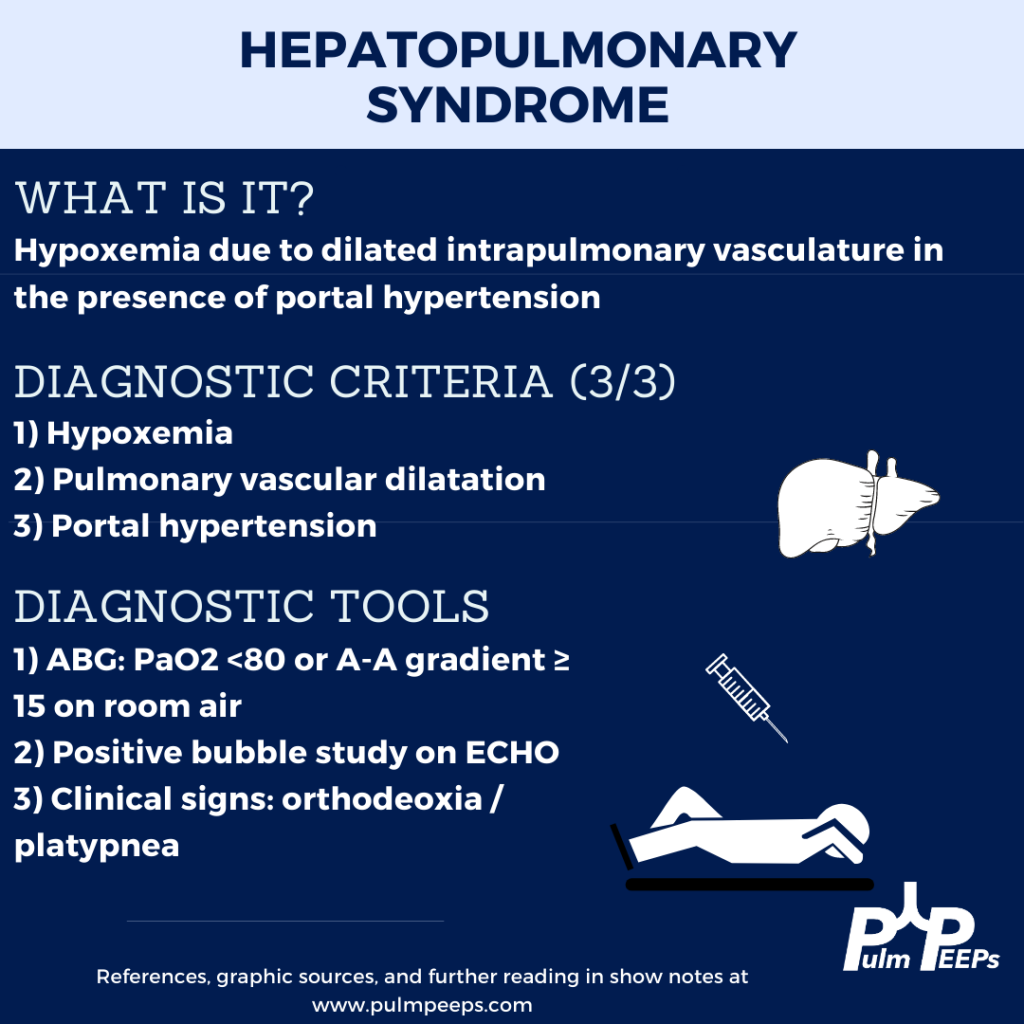
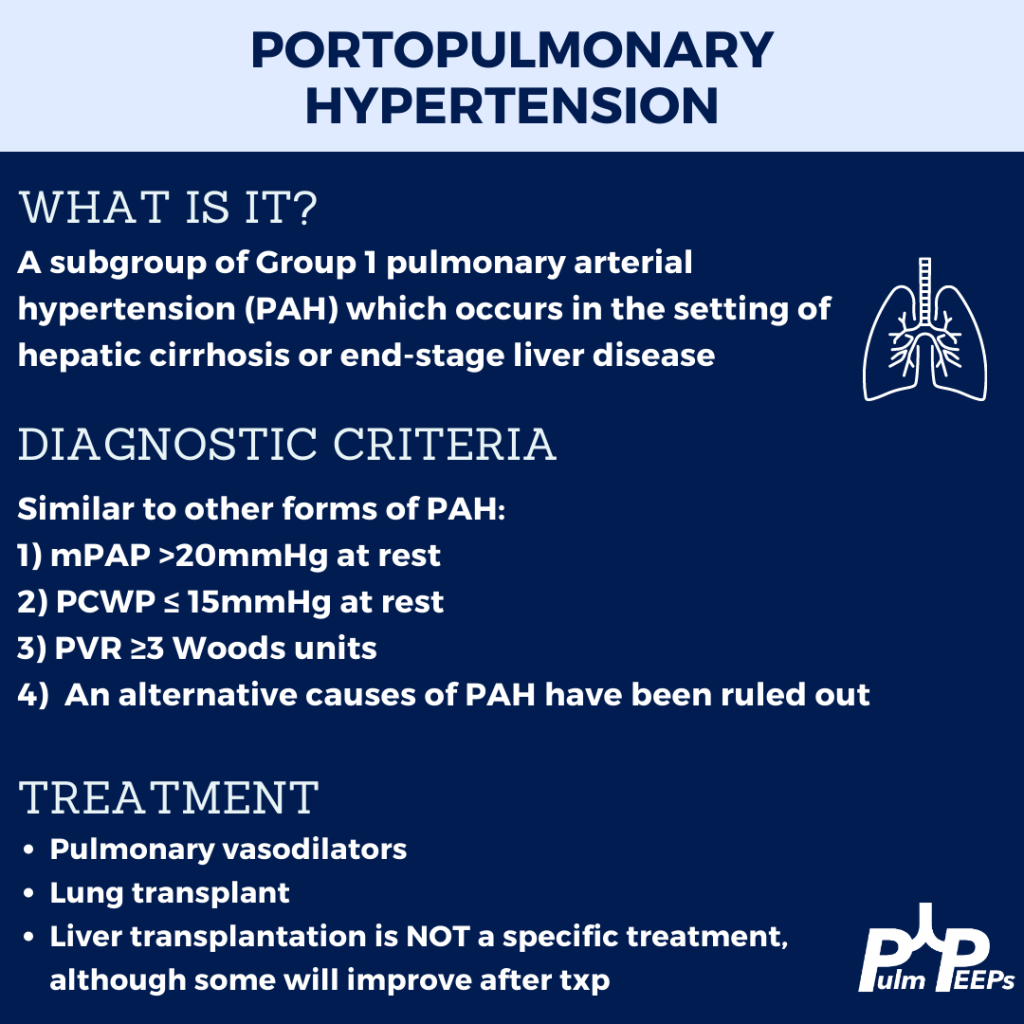
This week we are joined by one of our Associate Editors, Tess Litchman, as well as two guest experts to discuss two disease states that involve both the liver and the lung. Join us as we go through how to differentiate portopulmonary hypertension and hepatopulmonary syndrome.
Meet the Guests
Tess Litchman is a senior resident at Beth Israel Deaconess Medical Center and is one of the Associate Editors for PulmPEEPs. Tess will be continuing her training as a Pulmonary and Critical Care Medicine fellow at Brigham and Women’s Hospital next year.
Tyler Peck is an Instructor in Medicine at Beth Israel Deaconess Medical Center in the Division of Pulmonary and Critical Care Medicine. Tyler’s clinical and research interests are in pulmonary vascular disease and pulmonary hypertension.
Michael Curry is an Associate Professor of Medicine at Beth Israel Deaconess Medical Center and Section Chief of the Hepatology Department at BIDMC.
Further Readings and References
Rodríguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome–a liver-induced lung vascular disorder. N Engl J Med. 2008 May 29;358(22):2378-87. doi: 10.1056/NEJMra0707185. PMID: 18509123
Krowka MJ, Fallon MB, Kawut SM, et al. International Liver Transplant Society Practice Guidelines: Diagnosis and Management of Hepatopulmonary Syndrome and Portopulmonary Hypertension. Transplantation 2016; 100:1440.
Peppas, S., Nagraj, S., Koutsias, G., Kladas, M., Archontakis-Barakakis, P., Schizas, D., Giannakoulas, G., Palaiodimos, L., & Kokkinidis, D. G. (2022). Portopulmonary Hypertension: A Review of the Current Literature. Heart, lung & circulation, 31(9), 1191–1202. https://doi.org/10.1016/j.hlc.2022.04.056
This week on Pulm PEEPs, we are continuing our Top Consults series with a discussion on the work-up and diagnosis of Pulmonary Hypertension. See our prior Radiology Rounds on signs of PAH on CT scan, and listen to our follow-up episode on right heart catheterizations for some background before this episode… or dive right in! We’ll cover everything from history and physical, to recent guideline changes in the definition of PH, and much, much more!
Meet Our Guests
Erika Berman Rosenzweig is a Professor of Pediatrics and the Director of the Pulmonary Hypertension Center and CTEPH Program at Columbia University Medical Center / New-York Presbyterian Hospital. She is an active member of the Pulmonary Hypertension Association, was the Editor-in-Chief of Advances in Pulmonary Hypertension and is on the Scientific Board of the World Symposium on PH.
Catherine Simpson is an Assistant Professor of Medicine at Johns Hopkins Hospital and is one of the faculty members in our Pulmonary Hypertension group. Her clinical and research areas of expertise are in pulmonary vascular disease and right heart function. Her research is focused on novel biomarker discovery and metabolomics in pulmonary vascular disease.
Cyrus Kholdani is an Instructor in Medicine at Beth Israel Deaconess Medical Center and Harvard Medical School. He is also the director of the Pulmonary Hypertension Program at BIDMC, and is actively involved in clinical care and clinical research in a variety of pulmonary vascular disease domains.
Consult Patient
Ms. Pamela Harris (PH) is a 47-year-old woman with PMH of migraines, obesity s/p gastric sleeve (BMI now 33), and a history of remote DVT in her 20s while on OCP s/p 6 months of AC who is referred to pulmonary hypertension clinic for evaluation of dyspnea on exertion. She has actually had dyspnea for some time and previously it has been attributed to her weight. Based on this, she pursued a gastric sleeve and has lost 55 pounds, but continues to have shortness of breath. She has no cough, and does not get dyspnea at rest, but notes that after 1 flight of stairs, or 2-3 blocks on flat ground she has shortness of breath. She saw her PCP and had basic labs, basic spirometry, and an echocardiogram. He did not note anything significant on examination in the notes.
The labs had no anemia, and normal renal and liver function. Her serum bicarbonate was 25 and there was no blood gas. Spirometry showed an FVC 82% predicted, FEV1 83% predicted, and FEV1/FVC was 99% predicted. The echocardiogram had normal LVEF, mild LVH, normal RV size and function qualitatively. There was mild TR with tricuspid valve peak regurgitant velocity of 3.4 m/sec. The estimated PASP + RA pressure (based on normal IVC diameter 2.1 cm) was 46 mmHg.
RHC: Systemic BPs 140s/90s, with O2 saturations 97-98% on RA throughout. RA mean pressure was 9, RV was 48 with an RVEDP of 17, PA was 48/27 with mean of 34, and PCWP mean was 11. CO/CI by Fick was 5.56 / 2.42, and by thermo was similar, 5.8 / 2.52. Her PA sat was 62%, and PVR was 3.97 WU.
Key Learning Points
History
Understand the constellation of symptoms and the functional limitation
The goal is to assign a WHO functional class by the end of the visit
Evaluate the time course and evolution of the symptoms
Concerning symptoms that need to be addressed
Palpitations
Pre-syncope
Syncope
Chest pain
LE edema
Evaluate for risk factors to explain or contribute to pulmonary hypertension
Signs or symptoms of OSA
Signs or symptoms of auto-immune disease
Raynauds
Skin changes
Family history
Heritable lung disease
Clotting disorders
Auto-immune disease
Social history
Exposure history
Smoking
Physical Exam
Look for signs that confirm PH
Loud P2
Accentuated with elevated PVR
Can hear pretty early on. Could be one of the earliest findings
TR murmur – pansystolic murmur at RUSB
Diastolic murmur if severe pulmonary insufficiency
Look for signs of right heart failure
JVD
S4 gallop – later in course
RV heave – later in course
Peripheral edema
Pulsatile liver or hepatosplenomegaly
Look for signs of other secondary causes of PH
Mitral regurgitation or aortic stenosis murmur
Asymmetric lower extremity edema
Pulmonary edema
Skin findings concerning for auto-immune disease or liver disease
Arthritis
Work up for etiology of PH
CBC with diff – myeloproliferative and hemolytic anemia
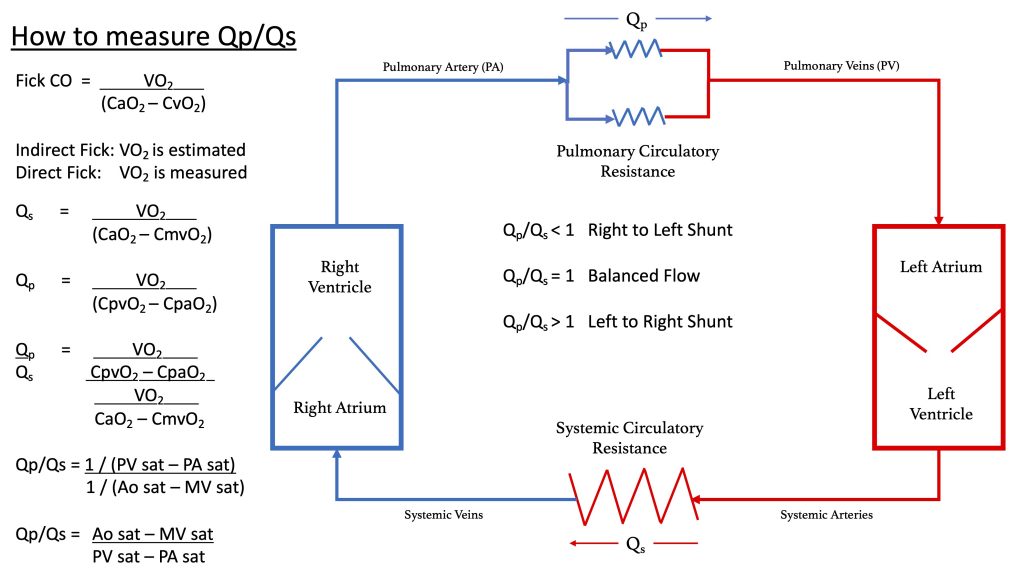
Today we have a special edition of Pulm PEEPs! We are revisiting our Radiology Rounds from 4 weeks ago to dive further into Right Heart Catheterizations and how to interpret them. We are joined by two experts in the field, Allison Tsao and Stephen Mathai.
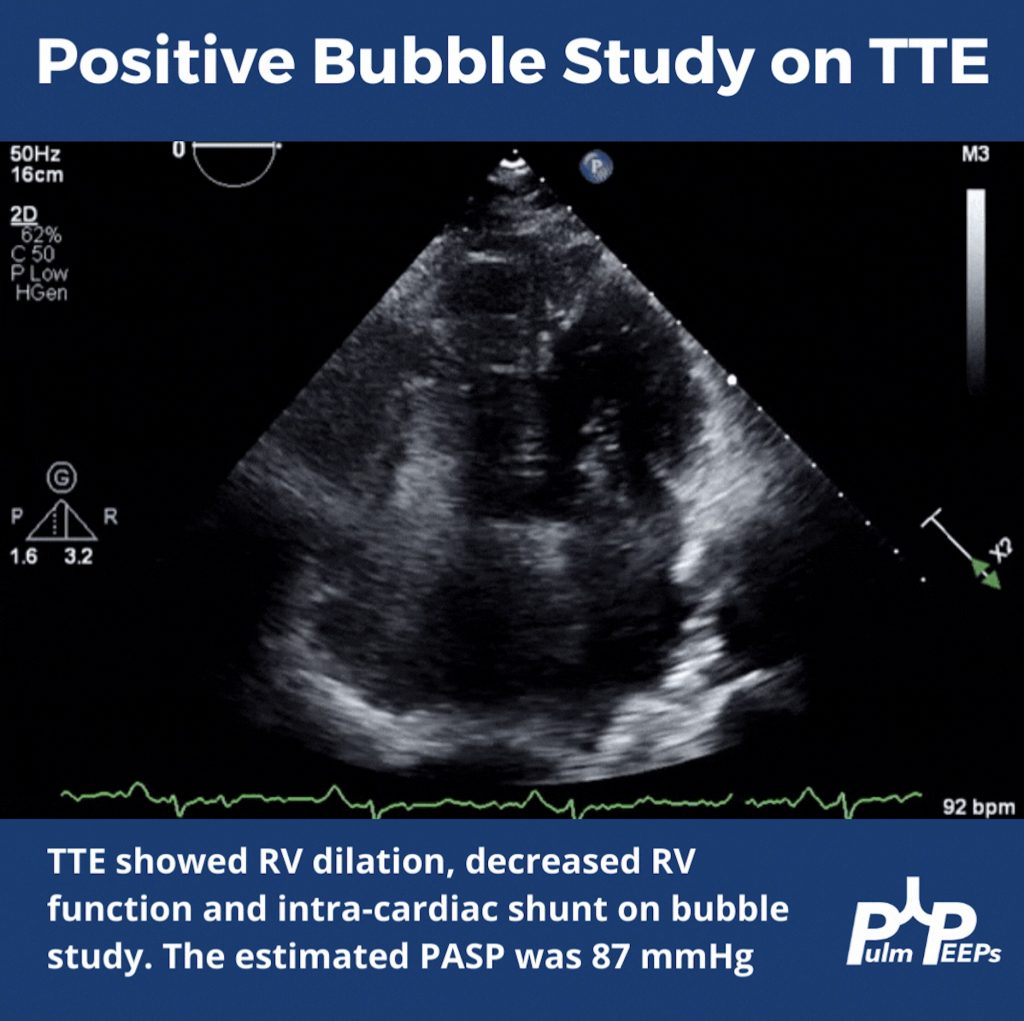
For a reminder, in that Radiology Rounds, we met a woman in her 50s with GERD, Raynaud’s, and multiple positive auto-antibodies (+ ANA 1:2560, + RNA pol III, + SSA, + anti-centromere) who presented with progressive dyspnea and was found to be hypoxemic. Her workup revealed severe pulmonary hypertension, and RV dysfunction on TTE with right to left shunting.
Meet Our Guests
Dr. Steve Mathai is an Associate Professor of Medicine at Johns Hopkins Hospital and the Director of the Inpatient Pulmonary Service. He specializes in Pulmonary Hypertension and his research focus is on scleroderma-associated PAH.
Dr. Allison Tsao is an Instructor in Medicine at Harvard Medical School and is an interventional cardiologist working at the Boston VA and Brigham and Women’s Hospital. She specializes in adult congenital heart disease and is the assistant director of the Translational Discovery Lab at BWH.