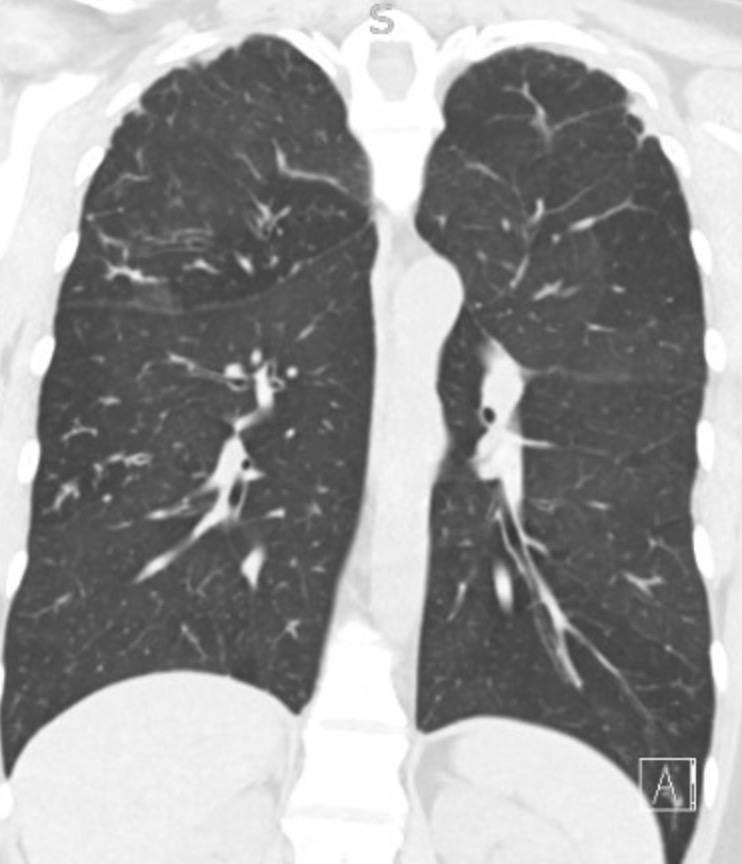
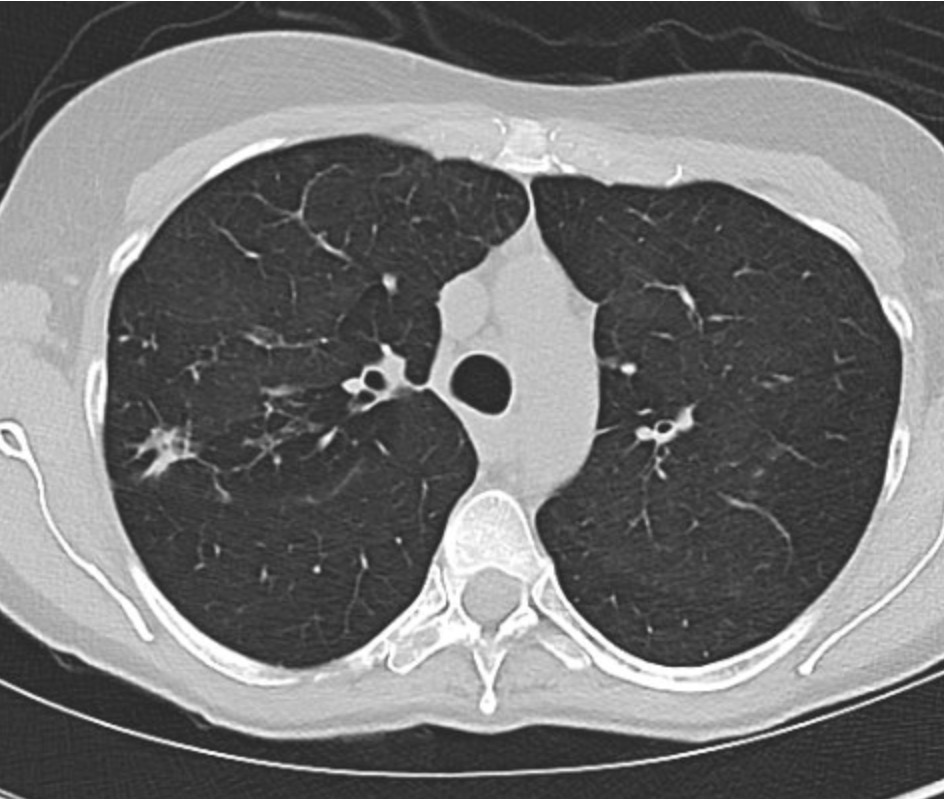
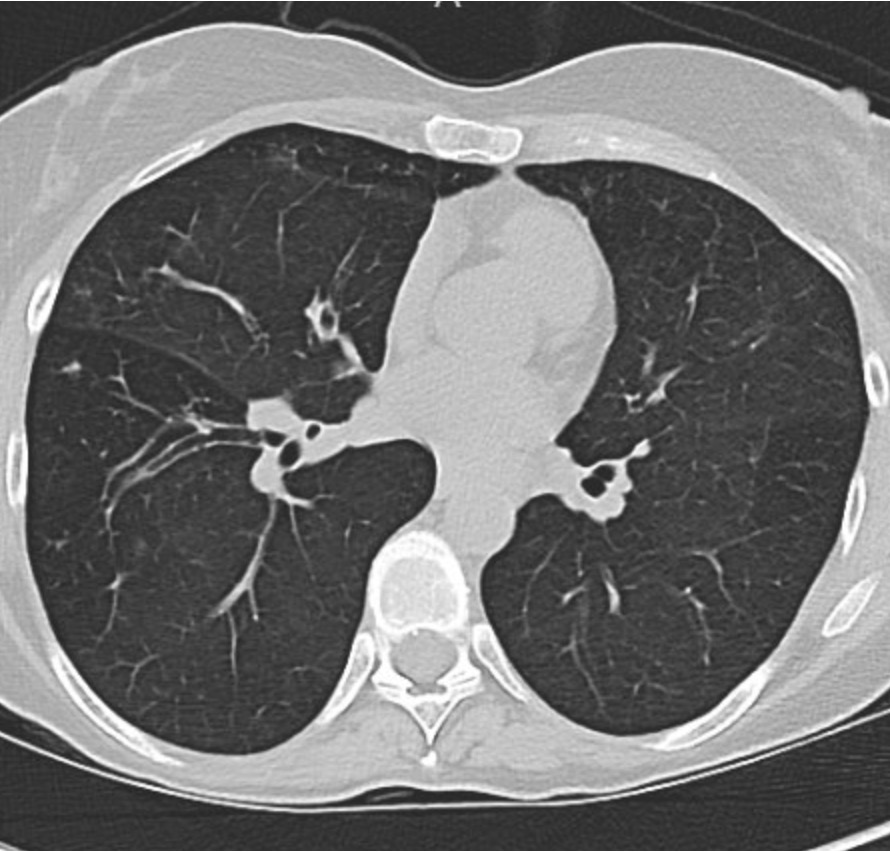
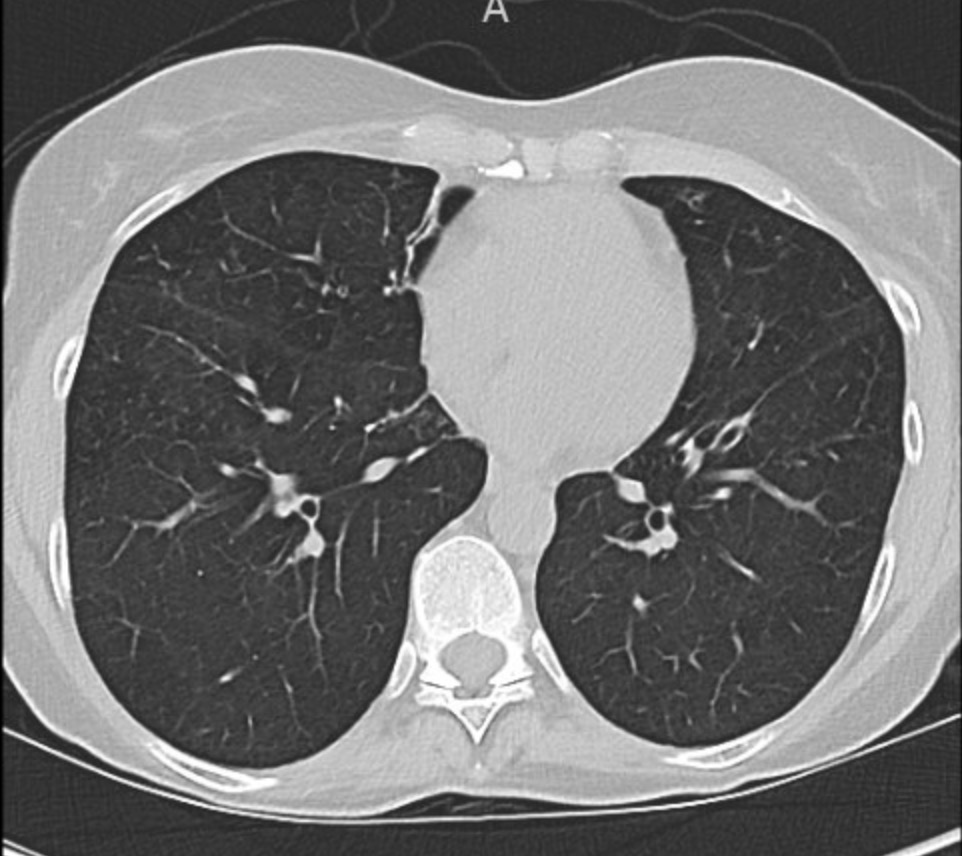
This week’s Pulm PEEPs Pearls episode is a focused discussion between Furf and Monty about non-pharmacologic techniques for airway clearance in the non-Cystic Fibrosis bronchiectasis population. This is a focused, high-yield discussion of the key points about airway clearance, including practical tips and a discussion of the evidence.
This episode was prepared in conjunction with George Doumat MD. Goerge is an internal medicine resident at UT Southwestern and joined us for a Pulm PEEPs – BMJ Thorax journal club episode. He is now acting as a Pulm PEEPs Editor for the Pulm PEEPs Pearls series.
Key Learning Points
1) Why airway clearance matters in non-CF bronchiectasis
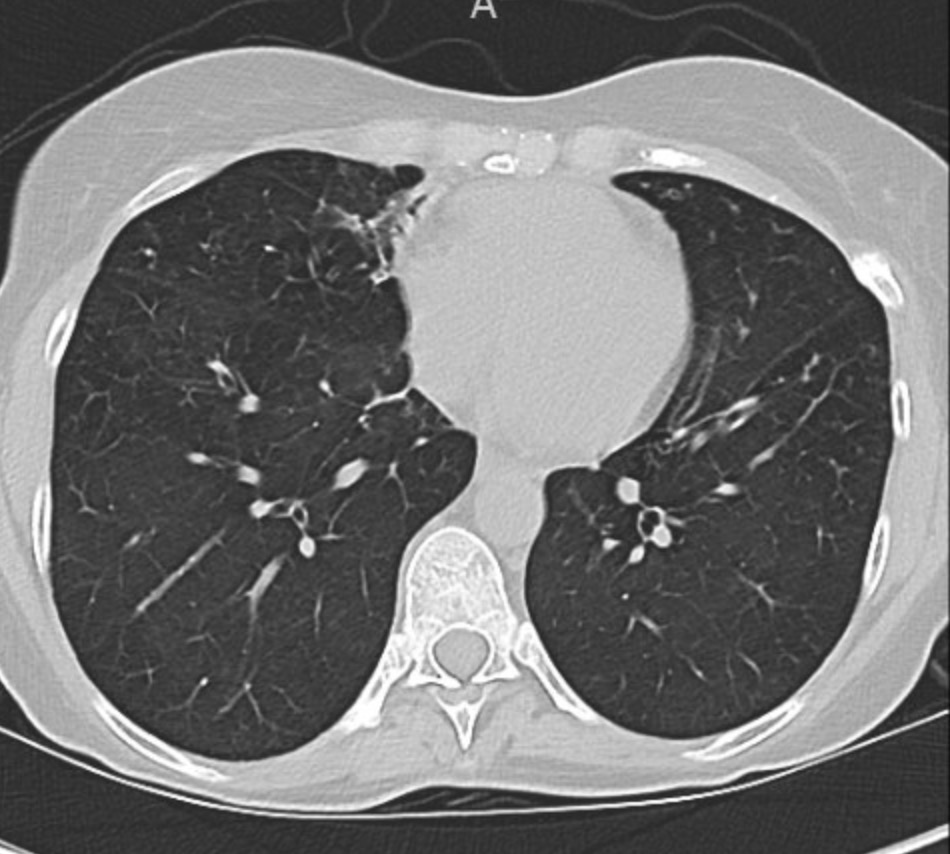
- Non-CF bronchiectasis is defined by irreversible bronchial dilation with impaired mucociliary clearance, leading to mucus retention.
- Retained sputum drives the classic vicious cycle: mucus → infection → neutrophilic inflammation → airway damage → worse clearance.
- Airway clearance techniques (ACTs) are meant to interrupt this cycle, primarily by improving mucus mobilization and symptom control.
2) What ACTs are trying to achieve clinically
- Main benefits are:
- More effective sputum clearance
- Reduced cough/dyspnea burden
- Improved activity tolerance and quality of life
- Effects on spirometry are usually small.
- Exacerbation reduction is possible, but evidence is mixed—some longer-term data suggest benefit for specific techniques.
3) The main ACT “families” and when to use them
Breathing-based techniques (device-free, flexible)
- ACBT (Active Cycle of Breathing Technique): breath control → deep breaths with holds → huffing.
- Pros: portable, adaptable, good first-line option.
- Key requirement: teaching/coaching to get technique right.
- Autogenic drainage: controlled breathing at different lung volumes to move mucus from peripheral → central airways.
- Pros: no device, can work well once learned.
- Cons: more technically demanding, needs training and practice.
PEP / Oscillatory PEP (stents airways + “vibrates” mucus loose)
- PEP: back-pressure helps prevent small airway collapse during exhalation; often paired with huff/cough.
- Oscillatory PEP (Flutter/Acapella/Aerobika): adds oscillation that many patients find easy and satisfying to use.
- Good fit for: people who benefit from airway stenting, want something portable, and prefer a device.
Mechanical/manual techniques (help when patient can’t self-clear well)
- HFCWO (“the vest”): external chest wall oscillation; helpful for high sputum volumes, dexterity limits, or difficulty coordinating breathing maneuvers.
- Postural drainage/percussion/vibration: caregiver/therapist-assisted options; still useful but consider:
- GERD/reflux risk with certain positions
- Hemoptysis risk with vigorous techniques
4) How to choose the “right” technique (the practical framework)
There is no one-size-fits-all. Match the tool to the patient:
- Sputum burden (volume/viscosity)
- Strength, coordination, cognition, dexterity
- Comorbidities (GERD, hemoptysis history, severe obstruction/airway collapse)
- Lifestyle + portability (what they’ll actually do)
- Cost/access and availability of respiratory therapy/physio support
A key mindset from the script: this is not a lifetime contract—reassess and adjust over time with shared decision-making.
5) Evidence takeaways (what improves, what doesn’t)
- ACTs reliably improve sputum expectoration and often symptoms/QoL.
- QoL/cough scores (e.g., SGRQ, LCQ) tend to improve modestly, particularly with oscillatory PEP and some vest studies.
- Lung function: typically minimal change; occasional short-term FEV₁ benefit is reported in some vest trials.
- Exacerbations: mixed overall; the script highlights a longer-term RCT of ELTGOL showing fewer exacerbations at 12 months vs placebo exercises.
- Safety: generally excellent; main cautions are hemoptysis and reflux (depending on technique/positioning).
6) Special population pearls
- Hemoptysis / fragile airways: start with gentle breathing-based ACTs (ACBT, controlled huffing); avoid overly vigorous oscillatory/manual methods if concerned.
- Severe obstruction or early airway collapse: PEP/oscillatory PEP can help by keeping small airways open on exhalation.
- Mobility/coordination barriers: consider HFCWO vest or simple oscillatory PEP devices to enable daily adherence.
- During exacerbations: keep it simple—1–2 reliable techniques, prioritize daily consistency, and re-check technique.
7) The “real” bottom line
- Start with simple, self-manageable options (often ACBT ± PEP).
- The “best” ACT is the one the patient will do consistently.
- Reassess technique and fit over time; education and demonstration are part of the therapy.
References and Further Reading
Lee AL et al., “Airway clearance techniques for bronchiectasis,” Cochrane Database Syst Rev. 2015; PMC7175838. PMID: 26591003.
Athanazio RA et al., “Airway Clearance Techniques in Bronchiectasis,” Front Med (Lausanne). 2020; PMC7674976. PMID: 33251032.
Iacono R et al., “Mucociliary clearance techniques for treating non-cystic fibrosis bronchiectasis,” Eur Rev Med Pharmacol Sci. 2015; PMID: 26078380.
Polverino E et al., “European Respiratory Society statement on airway clearance techniques in bronchiectasis,” Eur Respir J. 2023; PMID: 37142337.
Doumat G, Aksamit TR, Kanj AN. Bronchiectasis: A clinical review of inflammation. Respir Med. 2025 Aug;244:108179. doi: 10.1016/j.rmed.2025.108179. Epub 2025 May 25. PMID: 40425105.
Podcast: Play in new window | Download
Subscribe: Apple Podcasts | Spotify | Amazon Music | Android | iHeartRadio | Podcast Index | RSS